
使用螺旋桨HelixFold3预测生物分子结构
使用Paddle HelixFold3预测生物分子结构,有两种方式:
- 方式一:通过页面进行单次分子预测,适用于体验/测试场景;
- 方式二:通过OpenAPI进行批量分子预测,适用于生产环境;
背景
AlphaFold系列模型具备预测蛋白质单链、蛋白质复合体以及复杂生物分子结构的能力,凭借与实验方法相媲美的准确性,极大地推动了生命科学领域的进步。从AlphaFold2到AlphaFold-Multimer再到AlphaFold3,这些模型为科研工作者提供了强大的工具,显著提升了对蛋白质功能和相互作用的理解。尽管AlphaFold3支持生物分子相互作用的预测,技术本身对药物研发有极大的价值,但因其尚未开源被众多科学家所诟病,也阻碍了其在更广泛科研领域的应用和进一步发展。
完全复刻AlphaFold3的能力对促进生命科学领域的发展意义重大,但面临着模型结构和数据复杂性极高以及训练和运行所需的计算资源巨大等诸多挑战。在模型结构上,AlphaFold3 在 AlphaFold2 的基础上引入了原子级别的建模,使得整体建模更加复杂且计算量大幅增加。同时,AlphaFold3 使用了扩散模型来端到端地推理所有原子坐标。在数据层面上,由于 AlphaFold3 不仅需要建模蛋白质,还要处理小分子配体、核酸和离子等多种生物分子,复杂的数据预处理和大规模的自蒸馏数据生成对模型效果的增强至关重要。
百度螺旋桨 PaddleHelix 团队研发的 HelixFold3,在常规的小分子配体、核酸分子(包括 DNA 和 RNA)以及蛋白质的结构预测精度上已与 AlphaFold3 相媲美。具体效果指标参见:HelixFold3生物分子结构预测技术报告
使用方式一:通过页面进行单次分子预测
适用于体验/测试场景。
体验地址:https://paddlehelix.baidu.com/app/all/helixfold3/forecast
数据录入
输入实体
用户首先需要输入实体(Entity)数据。每一行为一条实体数据,您需要指定以下内容:
- 实体类型:目前我们支持的实体类型为“蛋白”、“配体”、“DNA”、“RNA”、“离子”
- 数量:代表该实体的复制数量,默认为1
- 序列:具体的实体内容。
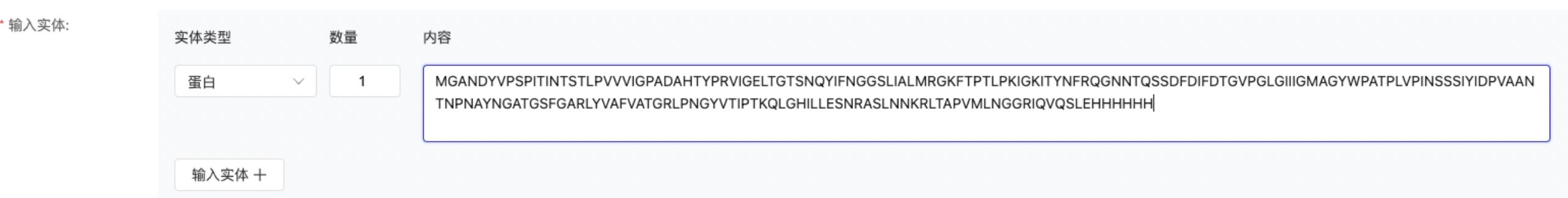
不同实体的输入要求与示例请参考下表:

^ 水、助剂和少量特殊的配体目前是模型所不支持的,我们会将这些配体从CCD列表中除去,如果您通过输入SMILES的方式进行了这些输入,可能会造成结果的表现下降。具体不支持的配体的CCD列表参见HelixFold3 FAQ。
请注意,除了上述输入要求,每个任务中所有实体加起来总token数量不能超过2000,不同实体的token计算方式参见HelixFold3 FAQ部分。
数据预览
- 实体类型为蛋白/DNA/RNA时,完成输入后,输入的序列会进行自动整理成为预览态。此时如果您需要编辑序列,直接点击序列即可重新恢复到编辑态
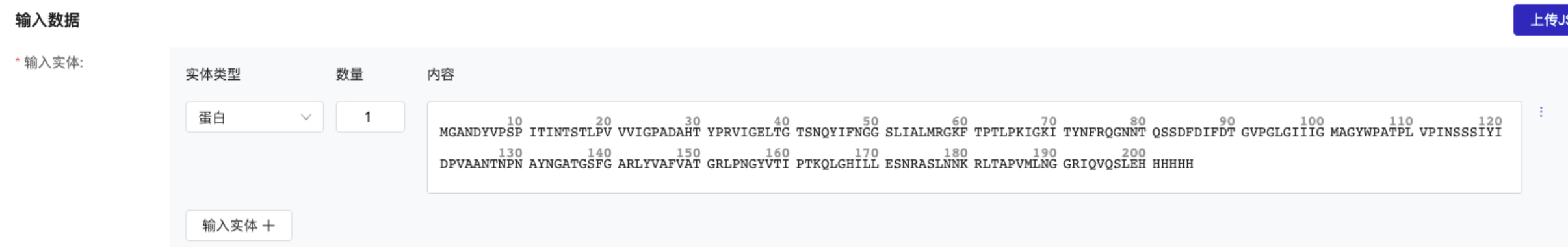
- 实体类型为配体时,完成输入后,输入框后方会出现“2D”标识。鼠标悬浮在这个标识上可以预览当前配体的二位结构。
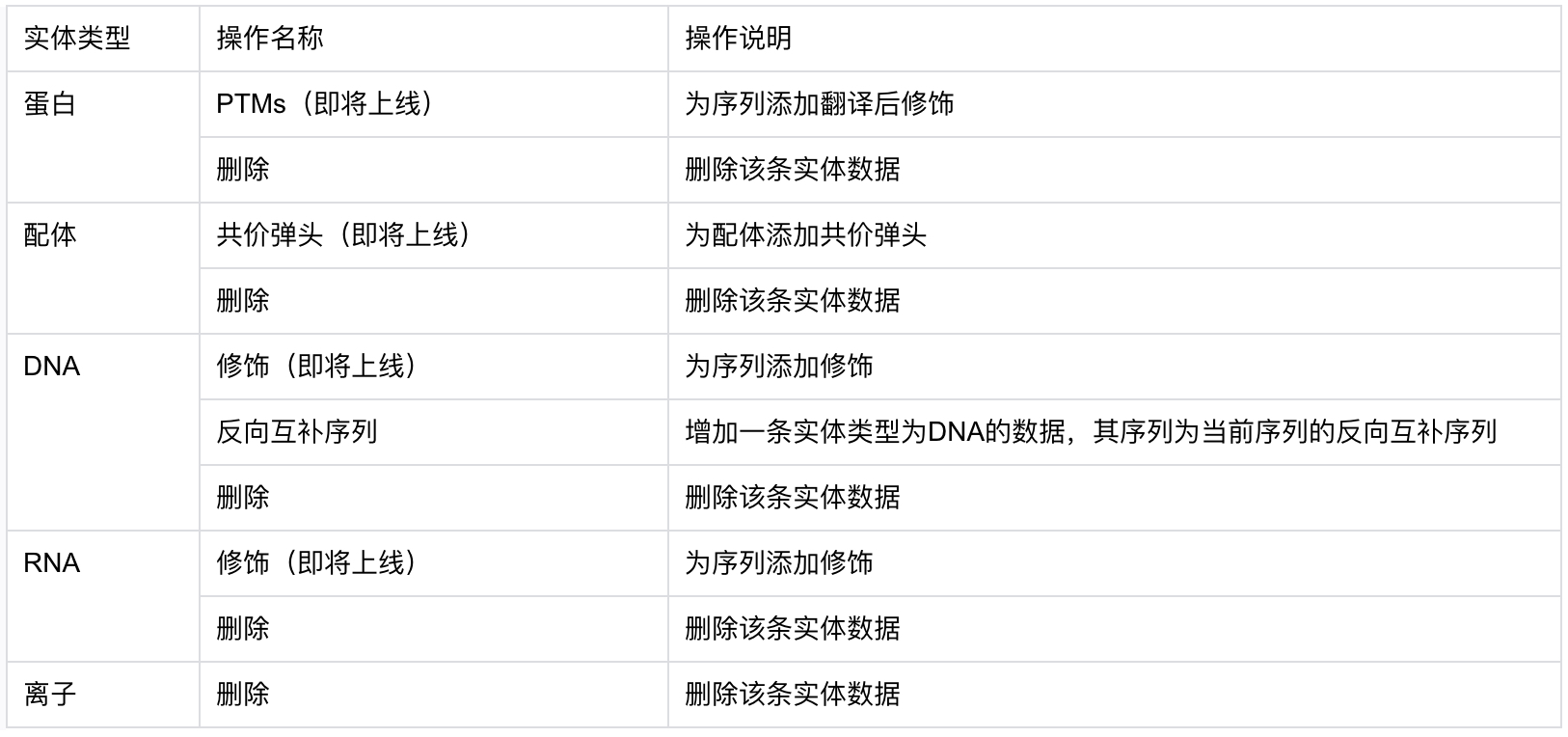
实体操作
点击每一行实体数据最后的三个点,可以对该条数据进行操作。以下是不同实体支持的操作一览:
指定相互作用(即将上线)
该模块支持用户指定已知的不同实体之间的相互作用,使得预测结果更贴合实际。
上传JSON
用户可以将需要预测的任务输入保存为JSON格式的文件,通过“上传JSON”按钮进行任务的快速读取和提交。JSON文件的说明与示例请参见:HelixFold3 JSON说明
查看结果
结果页呈现三个部分的信息:预测指标表格、PAE图和预测结构。
预测结果指标表格
左上角是预测指标表格,共四列:
- 第一列:结果的名称编号,每个任务默认输出5个结果
- 第二列:pLDDT (predicted local distance difference test)。这指的是局部置信度的每残差度量。它的范围从0到100,分数越高,表示置信度越高,通常预测也越准确。大于90的pLDDT被视为最高精度类别,其主链和侧链通常都能以高精度进行预测。相比之下,pLDDT高于70通常对应一些侧链错位的正确骨干预测。
- 第三列:pTM (predicted template modelling) 是一个综合指标,用于衡量模型对生物分子复合物整体结构预测的准确性。它表示的是预测结构与假设的真实结构叠加比对后的预估 TM 分数。TM 分数高于 0.5 表示预测的复合物整体折叠可能与真实结构相似,而 TM 分数低于 0.5 则意味着预测的结构可能是错误的。pTM 分数的定义与 TM 分数相同,但在解读 pTM 分数时需要谨慎。例如,在某些情况下,如果一个较大的蛋白结构被正确预测,而较小的伴随蛋白结构被错误预测,那么复合物的 pTM 分数可能会因为较大蛋白的贡献而高于 0.5。
- 第四列:ipTM (interface predicted template modelling)。ipTM 衡量的是生物分子复合物的亚基的相对位置的预测准确性。ipTM 值高于 0.8 表示高置信度的高质量预测,而低于 0.6 则表明预测可能失败。ipTM 值在 0.6 到 0.8 之间则属于灰色区域,预测可能是正确的,也可能是错误的。需要注意的是,即使复合物结构预测正确,ipTM 分数也可能因无序区域或 pLDDT 分数较低的区域而受到负面影响。ipTM 可能比 pTM 对用户更有用。这是因为亚基相对位置预测的质量与整个复合物预测的质量高度相关:如果亚基的相对位置是正确的(反映在较高的 ipTM 分数中),用户可以预期整个复合物结构也是正确的。
PAE图
您可以在此查看到预测排名第一的结构的PAE图。PAE (Predicted aligned error) 是一个二维图,蛋白残基沿着轴延伸。在每个方格中,绿色阴影表示一对残基的预期距离误差,单位为Ångströms(Å)。PAE图是衡量模型在预测结构内两个残基的相对位置上有多自信的指标。PAE被定义为当预测和实际结构在残基Y上对齐时,残基X处的预期位置误差,以Ångströms(Å)为单位测量。
来自两个不同域的两个残基之间的低PAE得分意味着低预测误差,也就是说,模型对这些残基的位置有信心。相反,PAE得分高意味着模型对其相对位置没有信心。
预测结构
您可以在3D视窗中查看预测排名第一的结构。
数据下载
所有5个结构的结果可以通过点击“下载结果”按钮来下载。下载好的压缩包的层级结构如下
1task_name.zip/
2│
3├──user_input.json
4│
5├──*-rank1/
6│ ├─all_results.json
7│ └─predicted_structure.cif
8│
9├──*-rank2/
10│ ├─all_results.json
11│ └─predicted_structure.cif
12│
13├──*-rank3/
14│ ├─all_results.json
15│ └─predicted_structure.cif
16│
17├──*-rank4/
18│ ├─all_results.json
19│ └─predicted_structure.cif
20│
21└───*-rank5/
22 ├─all_results.json
23 └─predicted_structure.cif- user_input.json- 本次任务输入数据的json文件。可以用于重新提交任务或回顾本次任务的输入。
-
*-rankX/- 包含排序为第X的预测结果的文件夹
- all_results.json- 该预测结果的相关指标。具体包含的内容如下
- predicted_structure.cif- 该预测结果的结构文件
使用方式二:通过OpenAPI进行批量分子预测
适用于生产环境。
目前限制白名单使用,如需使用请工单联系开通。
接口说明
准备工作
接口描述
该接口可用于提交和查看HelixFold任务。
接口鉴权
- 百度云如何对您的API请求进行鉴权
当您将HTTP请求发送到百度智能云时,您需要对您的请求进行签名计算,以便百度智能云可以识别您的身份。您将使用百度智能云的访问密钥来进行签名计算,该访问密钥包含访问密钥ID(Access Key Id, 后文简称AK)和秘密访问密钥(Secret Access Key, 后文简称SK).
了解如何创建、查看和下载Access Key Id(AK)和Secret Access Key(SK), 请参考如何获取AKSK - 签名API请求
在请求签名之前,请先计算请求的哈希(摘要)。然后,您使用哈希值、来自请求的其他信息以及您的秘密访问密钥(Secret Access Key,SK),计算另一个称为签名(Signature)的哈希, 得到签名后,进行一定规则的拼装成最终的认证字符串,也就是最终您需要包含在API请求中的Authorization字段。
您可以通过以下方式携带认证字符串:
- 在HTTP Header中包含认证字符串
- 在URL中包含认证字符串用户也可以将认证字符串放在HTTP请求Query String的authorization参数中。常用于生成URL给第三方使用的场景,例如要临时把某个数据开放给他人下载。关于如何在URL中包含认证字符串,请参考在URL中包含认证字符串。
您可以参看从零开始用Python调用API接口,视频的前半部分介绍了百度智能云鉴权认证机制,帮助您更快的进行了解。
- 使用 Python 进行请求的鉴权代码样例
1#!/usr/bin/env python3
2# -*- coding: utf-8 -*-
3
4
5import hashlib
6import hmac
7import urllib
8import time
9
10# 1.AK/SK、host、method、URL绝对路径、querystring
11AK = "input your AK"
12SK = "input your SK"
13host = "chpc.bj.baidubce.com"
14method = "POST"
15query = ""
16URI = "/api/submit/helixfold"
17
18# 2.x-bce-date
19x_bce_date = time.gmtime()
20x_bce_date = time.strftime('%Y-%m-%dT%H:%M:%SZ',x_bce_date)
21# 3.header和signedHeaders
22header = {
23 "Host":host,
24 "content-type":"application/json;charset=utf-8",
25 "x-bce-date":x_bce_date
26 }
27signedHeaders = "content-type;host;x-bce-date"
28# 4.认证字符串前缀
29authStringPrefix = "bce-auth-v1" + "/" +AK + "/" +x_bce_date + "/" +"1800"
30# 5.生成CanonicalRequest
31#5.1生成CanonicalURI
32CanonicalURI = urllib.parse.quote(URI) # windows下为urllib.parse.quote,Linux下为urllib.quote
33#5.2生成CanonicalQueryString
34CanonicalQueryString = query # 如果您调用的接口的query比较复杂的话,需要做额外处理
35#5.3生成CanonicalHeaders
36result = []
37for key,value in header.items():
38 tempStr = str(urllib.parse.quote(key.lower(),safe="")) + ":" + str(urllib.parse.quote(value,safe=""))
39 result.append(tempStr)
40result.sort()
41CanonicalHeaders = "\n".join(result)
42#5.4拼接得到CanonicalRequest
43CanonicalRequest = method + "\n" + CanonicalURI + "\n" + CanonicalQueryString +"\n" + CanonicalHeaders
44# 6.生成signingKey
45signingKey = hmac.new(SK.encode('utf-8'),authStringPrefix.encode('utf-8'),hashlib.sha256)
46# 7.生成Signature
47Signature = hmac.new((signingKey.hexdigest()).encode('utf-8'),CanonicalRequest.encode('utf-8'),hashlib.sha256)
48# 8.生成Authorization并放到header里
49header['Authorization'] = authStringPrefix + "/" +signedHeaders + "/" +Signature.hexdigest()
50# 9.发送API请求并接受响应
51import requests
52import json
53
54body={
55 "name" : "QQQQQQ"
56 }
57
58url = "http://"+host + URI
59
60r = requests.put(url,headers = header,data=json.dumps(body))
61
62print(r.text)接口信息
helixfold 作业提交

请求参数:
1{
2 "job_name": "Protein-RNA-Ion: PDB 8AW3", # string | 任务名,如未指定,则系统自动生成
3 "recycle": 4, # 可选,int | 范围[1,100]。模型推理参数,影响模型效果,越大越好。如果用户不设置,默认设为4。
4 "ensemble": 1, # 可选,int | 范围[1,100]。模型推理参数,影响模型效果,越大越好。如果用户不设置,默认设为1。
5 "entities": [ # list | 定义各种实体。支持的实体类型:“protein”,“dna”,“rna”,“ion”,“ligand”。注:每个任务中所有实体加起来总token数量不能超过2000,不同实体的token计算方式见下方注释或参见HelixFold3 FAQ部分。
6 {
7 "type": "protein", # string | 实体类型(此处以蛋白为例)
8 "sequence": "GPDSMEEVVVPEEPPKLVSALATYVQQERLCTMFLSIANKLLPLKP", # string | 蛋白序列,仅支持20种标准氨基酸。一个氨基酸算做一个token,最大不可超过2000。
9 "count": 1 # int | 实体复制数量
10 },
11 {
12 "type": "ion", # string | 实体类型(此处以离子为例)
13 "ccd": "ZN", # string | 离子的CCD标准名字。目前支持的离子列表请参考“离子CCD列表”。一个离子算作一个token。
14 "count": 2 # int | 实体复制数量。实体为离子时,数量不可超过50。
15 },
16 {
17 "type": "dna", # string | 实体类型(此处以DNA为例)
18 "sequence": "ACGTTTACGGGG", # string | DNA序列,仅支持4种标准脱氧核糖核酸ATCG。一个核苷酸算做一个token,最大不可超过2000。
19 "count": 1 # int | 实体复制数量
20 },
21 {
22 "type": "ligand", # string | 实体类型(此处以配体为例)
23 "ccd": "ATP", # string | 支持用户用CCD输入小分子配体,CCD和下方的SMILES只需输入其中一个即可。如果两个字段中都有输入,则以CCD的输入为准。配体中的一个原子算做一个token。注:水、助剂和少量特殊的配体目前是模型所不支持的,我们会将这些配体从CCD列表中除去,如果您通过输入SMILES的方式进行了这些输入,可能会造成结果的表现下降。具体不支持的配体的CCD列表参见[HelixFold3 FAQ]。
24 "smiles": "CCccc(O)ccc", # string | 小分子的SMILES,重核数量需在50以内。SMILES和上方的CCD只需输入其中一个即可。如果两个字段中都有输入,则以CCD的输入为准。配体中的一个原子算做一个token。
25 "count": 1 # int | 实体复制数量。实体为配体时,数量不可超过50。
26 },
27 {
28 "type": "ion", # 同上离子部分。
29 "ccd": "ZN",
30 "count": 1
31 },
32 {
33 "type": "dna", # 同上 dna 部分。
34 "sequence": "CCCCGTAAACGT",
35 "count": 1
36 },
37 {
38 "type": "rna", # string | 实体类型(此处以RNA为例)
39 "sequence": "ACCCCCCC", # string | 序列,仅支持4种标准核糖核酸AUCG。一个核苷酸算做一个token,最大不可超过2000。
40 "count": 1 # int | 实体复制数量。
41 }
42 ]
43}
返回参数
1{
2 "code": 0, # int | 状态码
3 "msg": "xxx", # string | 提示信息
4 "data": {
5 "task_id": 123 # uint64 | 任务ID
6 }
7}helixfold 任务结果查询
请求参数
1{
2 "task_id": 123 # uint64 | 任务ID
3}
返回参数
1{
2 "code": 0, # int | 状态码
3 "msg": "xxx", # string | 提示信息
4 "data": {
5 "status": 10, # 任务执行状态
6 "run_time": 123, # 任务执行时间
7 "result": "http:xxx", # 结果临时下载URL
8 }
9}
HelixFold3使用条款与许可证请参考:HelixFold3 Server 使用条款
评价此篇文章